- General information
- What you can do with RNA FRABASE
- Search
- Basic search
- Input data format (sequence & structure)
- Examples (sequence & structure)
- Advanced search
- Loading input data from file
- Saving input data
- Structural elements
- Residue
- Base pair
- Multiplet
- Dinucleotide Step
- Stem
- Loop
- Results
- Atom coordinates for selected fragment(s)
- Torsion and pseudotorsion angles, pseudorotation, sugar pucker amplitude for selected fragment(s)
- Base pair classification for selected fragment(s)
- Secondary structures
- How you can cite RNA FRABASE
Here, we present RNA FRABASE in its 2.0 version. This is an advanced tool which should greatly
facilitate the RNA structure analysis and modelling. In comparison to RNA FRABASE 1.0 new functionalities (menu
Structural elements and
Secondary structures)
as well as a new data, were introduced.
If one compares the three dimensional RNA structure to a spatial puzzle, RNA FRABASE allows to pull out a defined piece of this puzzle - the 3D RNA fragment.
The architecture of the web-accessible RNA FRABASE engine and database is based on the following information
path:
PDB-deposited RNA structures |
→ |
RNA sequences & secondary structures described in the dot-bracket notation |
→ |
secondary structures of RNA fragments |
→ |
3D RNA fragments |
The database contains:
- RNA sequences and secondary structures (described in the dot-bracket notation and presented in a graphic form)
derived from the PDB-deposited RNA structures and their complexes;
- atom coordinates of the unmodified and modified nucleotide and nucleoside residues
taken from the PDB-deposited RNAs and their complexes;
- calculated torsion and pseudotorsion angle values, sugar pucker parameters and complete classification of base pair types.
- calculated base-base parameters for base pairs and inter base pair parameters for dinucleotide steps;
to be applied as filters when searching these structural elements
Current state of the database is reported on the
Search page, where one can find information about:
- the date of the recent database update with new structures;
- the number of RNA structures and their complexes included in the database;
- the number of atom coordinates for residues included in the database.
To create an entry in the database, we extract sequence(s) and atom coordinates of nucleotide residues from Protein Data Bank.
Secondary structure for each RNA molecule is reconstructed by our script using
RNAView software
(Yang, H., Jossinet, F., Leontis, N., Chen, L., Westbrook, J., Berman, H., Westhof, E. (2003)
Tools for the automatic identification and classification of RNA base pairs.
Nucleic Acids Research, 31, 3450-3460)
and a function encoding the structure in the dot-bracket notation.
All sugar, backbone and χ torsion and pseudotorsion angles are calculated using our own scripts. Base pair classification is based on
the RNAView software. Base-base parameters for base pairs and inter base pair parameters for dinucleotide steps were
calculated using
3DNA software (Lu,X.-J., Olson,W.K. (2003)
3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures.
Nucleic Acids Research, 31, 5108-5121).
Every database entry is automatically checked for conformational errors.
RNA FRABASE is served by Apache http daemon along with the PHP scripts. It runs on SUSE Linux. PostgreSQL is used as a relational database.
RNA FRABASE was tested using following browsers: Opera 10, Mozilla Firefox 3.6, MS Internet Explorer 6/7/8, Google Chrome 4 and Apple Safari 4.
The RNA tertiary structure is formed by folded single oligoribonucleotide strand.
RNA FRABASE is designed to search for user-specified, RNA fragments
which may be single or multi-stranded (c.f. figure below).
The database input needs to specify those strands.
See below for the secondary structure visualization od 5S rRNA and its
requested fragment using
PseudoViewer software (Byun,Y., Han,K. (2009)
PseudoViewer3: generating planar drawings of large-scale RNA structures with pseudoknots.
Bioinformatics, 25, 1435-1437).
Search for RNA fragment(s) is performed after submitting one of the following types of query:
- basic search
user defines:
- RNA sequence(s);
- secondary structure(s) of the fragment;
- advanced search (i.e. basic search with filtering options)
user specifies:
- modified residues sensitive search;
- through-space interactions;
- analysis of all NMR models of the structure;
- strand shift operation;
- experimental method used for structure determination;
- resolution;
- limits for conformational parameters (pseudorotation, torsion and pseudotorsion angles, sugar pucker amplitude);
- structural elements.
RNA FRABASE search engine finds all three-dimensional RNA fragments
exactly matching user specification.
As a result of query the search engine returns the
list of suitable RNA fragments
which can be subjected to an instant visualization using the
JSmol software.
For each 3D fragment from the list the following structural parameters are available:
All of these parameters can be saved and used for further analysis.
Search page contains the following functional objects:
>strand1
GAGGNRACUC
(((....)))
|
sequence / structure entry box (enter the query in the appropriate format here) |
|
| Load example: 1 2 3 4 5 6 |
click a number to load one of the 6 example queries into the sequence / structure entry box |
|
| C:/my_input.txt |
Browse |
click this button to browse file with sequence / structure entry |
|
| Load file |
|
click this button to load sequence(s) / structure(s) from selected file |
|
| Save |
|
click this button to save sequence(s) / structure(s) from the sequence / structure entry box to the file |
|
| Reset |
|
click this button to clear the sequence / structure entry box |
|
| Search |
|
click this button to run RNA FRABASE search engine |
|
| Advanced Search |
click this button to open the page with advanced settings |
To facilitate searching, six
examples were provided.
A new user may start his/her adventure with the RNA FRABASE looking at them
(the pictorial representation of the structure described in an example can be seen when one points
at the appropriate example's number) and running the search with any
example uploaded. In further work, the examples may serve as templates of basic query.
Obligatory format of the basic query is described in the following section.
A single search concerns one RNA fragment which can be single- or multi-stranded.
Number of strands which compose the fragment is
unlimited (e.g. seven-way junction structure can be queried in the database).
Every strand should be defined in one separate section. Section concerning a strand may be composed of two or three lines:
| line |
type |
contents |
| 1st line |
obligatory |
">" followed by a unique identifier of the strand (composed of <= 8 alpha-numeric characters) |
| 2nd line |
optional |
RNA sequence in the one-letter format coherent with IUPAC Code |
| 3rd line |
optional for single-stranded fragments |
secondary structure in the dot-bracket notation |
(Optionally, in any place of the entry, user can insert a comment line starting from "#".)
First line indicates where the section begins and it must be placed before the specification of a sequence and secondary structure.
This line is followed by the exact definition of the strand, in which user can specify:
- only the sequence;
- only the secondary structure;
- the sequence and the secondary structure.
In the first case (
only sequence is defined), sequence should be composed of at least 4 letters from the {A, C, G, U} set
(in the case of longer sequences all the letters from
IUPAC Code table are accepted).
This lower bound has been set because of computational matter (the number of fragments matching e.g. 'CG' query (or any sequence containing only 2 residues)
is huge, which makes further work with the results hardly possible, and the search occupies a lot of computational power).
In the second case (
only secondary structure is defined), lower bound for the number of characters is set for the same reasons
as above. At least 6-11 characters should be specified (number of characters depends on the character types and is computed
by the lower_bound weighted function; 6 characters are enough when secondary structure is encoded with the brackets only; 11 characters
are the smallest number when only the dots are used to encode the structure).
In the third case (entry contains both
sequence and secondary structure description), the amount of characters in both lines
must be the same. Lower bound for the number of characters in each description is also set.
When searching for fragments containing
two or more strands the following conditions must be fulfilled:
- each strand must be connected with the subsequent one at least by one W-C base pair or GU,
- the final strand must be connected with the first one,
- the number of opening and closing brackets should be the same for the whole fragment.
The following characters are accepted when entered into the
sequence / structure entry box:
| type of contents | accepted characters (wildcards) |
| strand identifier | >, A-Z, a-z, 0-9 |
| sequence | A, C, G, U, R, Y, M, K, W, S, B, D, H, V, N |
| secondary structure | . ( ) [ ] { } < > ? |
| sequence and / or secondary structure | ^ $ |
| comment line | # - as the first character in line |
Search engine ignores:
- empty lines;
- comment lines, i.e. lines starting from "#",
if placed in the
sequence / structure entry box.
The sequence should be encoded with characters from the set defined by
IUPAC Nucleic Acid Code for Ribonucleosides.
Both upper and lower case letters are accepted.
In the case of the basic search, RNA FRABASE engine does not differentiate between upper and lower case characters entered by the user.
In the case of the advanced search, when the
modified residues sensitive search option is checked the following rule is used:
| UPPER CASE CHARACTER | - represents non-modified ribonucleotide residue |
| lower case character | - represents modified ribonucleotide residue (e.g. deoxy) and 5'-dephosphorylated residues |
IUPAC Nucleic Acid Code determines the manner of encoding any subset
of four nucleotides with one-letter symbol. The following table
contains the complete list of the symbols used to encode ribonucleosides and the corresponding subsets.
| Code | Description |
| A | Adenosine |
| C | Cytidine |
| G | Guanosine |
| U | Uridine |
| R | Purine ribonucleoside {A, G} |
| Y | Pyrimidine ribonucleoside {C, U} |
| M | {C, A} |
| K | {U, G} |
| W | {U, A} |
| S | {C, G} |
| B | {C, U, G} (not A) |
| D | {A, U, G} (not C) |
| H | {A, U, C} (not G) |
| V | {A, C, G} (not U) |
| N | any ribonucleoside {A, C, G, U} |
In the dot-bracket notation:
- an unpaired nucleotide is represented as a dot "."
- a base pair is represented as a pair of opening (left) and closing (right) brackets, i.e. "(" and ")".
This notation is extended to represent secondary structure of pseudoknots,
by inserting squared "[" and "]" brackets for lower-order structures.
The curly brackets "{" and "}" and angle brackets "<" and ">"
are used for higher and most complicated pseudoknots.
- a missing residue is represented as a dash "-"
-
Following wildcards were introduced to the search pattern:
- "^" (up arrow) determines search for the 5'-end fragment only
- "$" (dollar sign) determines search for the 3'-end fragment only
- "?" (question mark) stands for any residue which might be paired or unpaired.
Wildcards "^" and "$" can be placed only at the beginning or at end of the strand both within a single or multistrand fragments of interest.
Wildcard "?" can be placed in any place but only for single strand fragments. In case of multistrand
fragments this wildcard operates only on the 5'- and 3'-ends of the respective strand (see examples below).
Example 1
|
Example 2
|
Example 3
|
Example 4
|
Example 5
|
Example 6
|
Six examples, ready to be loaded, are available to a user (see section
Basic search).
Examples 1 and 2 concern searching for single-stranded fragments (tRNA, hairpin loop), the remaining ones refer to multi-stranded fragments.
The following table shows graphical representation of each example (except from example 1) and the corresponding RNA FRABASE entry.
Example 1
#tRNA(Phe), yeast (Saccharomyces Scerevisiae)
>strand1
gCGGAUUUAgCUCAGuuGGGAGAGCgCCAGAcUgAAgAuc
UGGAGgUCcUGUGuuCGaUCCACAGAAUUCGCACCA
|
Example 2
|
Example 3
|
Example 4
|
Example 5
|
Example 6
|
Optionally a user can run the search with advanced options specified. To enter
Advanced search page one should first define basic search (i.e. specify sequence and / or secondary structure on the main
Search page) and then click
Advanced Search button.
Advanced search provides the following possibilities:
- Search for RNA structures containing modified ribonucleotide residues
(to do so, activate the modified residues sensitive search check-box. By default this option is not checked and the search engine does not differentiate between upper- and lower-case characters in the queried sequence);
- Add through-space interactions: by default this option is not checked and the search engine follows exactly
the secondary structure input pattern. When active, all loop residues are scanned, both those represented by
"." and those represented by brackets. This allows to find through-space interactions in the higher-order
structures like kissing loops and pseudoknots
- Include all models of the structure: to do so, activate the check-box. By default this option is not
checked and the search engine presents only the model No.1.
- Strand shift operation
: this allows for considerable enlargement of the RNA di- and multistrand- fragments
population. By default this option is not checked and the search engine follows exactly the RNA FRABASE secondary
structure input pattern. This pattern describes which strand is the first one in the PDB structure. When active,
this priority is shifted to the subsequent strands (i.e. an analogy to a bitwise shift operator). The idea is
presented below in a case of the three-way junction. For this example when the strand shift operation is OFF no fragment is
found and when it is checked ON - six fragments are found.
- Search with the following filters:
- the experimental method used to determine RNA structure and its resolution, (to activate the
filter select the appropriate check-box and type the resolution value in the resolution entry box);
- ranges of the conformational parameters (torsion and pseudotorsion angle values, sugar pucker parameters)
(type limit ranges in the limits entry box).
Limits for conformational parameters should be typed in the appropriate format, which is described in the following section.
There is a variety of possibilities to define limits for values of conformational parameters in
the
limits entry box.
User may define the limits for:
- one or more selected strand(s);
- one or more selected residue(s) within the strand;
- one or more selected conformational parameter(s);
A single line typed in the
limits entry box should have the following format:
| <residue number> <parameter symbol> <min value> <max value> |
Thus,
one line concerns the limit for a
single conformational parameter, which
should be satisfied in
a single or all residue(s) within the requested RNA fragment. For example:
| the following line | means that we search for... |
|
| 2 alpha 120 180 |
the fragment having: in residue number 2, parameter alpha in the range <120, 180> |
|
| all vmax 30 50 |
the fragment having: in all the residues, parameter vmax in the range <30, 50> |
How can a user find the proper numbering of residues within the strand(s) of the fragment?
The RNA FRABASE makes this quite simple:
- When a user defines the sequence in the sequence / structure entry box and clicks
Advanced Search button (see section Basic Search), all the residues within the defined strand(s) are numbered
in the automatic way.
- Next, a user enters the Advanced Search page and has the limit template loaded into
the limits entry box.
- A user should modify the template according to his/her interests.
The template, loaded into the
limits entry box
concerns the
queried sequence.
It contains a list of commented lines divided into two sections:
- First section of the template, informs about:
- line format,
- the strands specified by a user, each strand name is given in a separate comment line,
- numbering of residues for each strand.
- Second section contains the list of conformational parameters and their limits.
The following table shows example templates placed by default into the
limit entry box, when
a user decides to make an advanced search for
Example 1 and for
Example 3.
By default, all the parameters have value within the range <-180, 180> and all the lines are commented ("#" placed as the first character in each line).
| limit template for Example 1 |
limit template for Example 3 |
#<no> <angle> <min> <max>
#Strand named: strand1
#residue numbers: 1-76
#all alpha -180 180
#all beta -180 180
#all gamma -180 180
#all delta -180 180
#all epsilon -180 180
#all zeta -180 180
#all chi -180 180
#all p -180 180
#all vmax -180 180
#all v0 -180 180
#all v1 -180 180
#all v2 -180 180
#all v3 -180 180
#all v4 -180 180
#all eta -180 180
#all theta -180 180
|
#<no> <angle> <min> <max>
#Strand named: strand1
#residue numbers: 1-6
#Strand named: strand1
#residue numbers: 7-12
#all alpha -180 180
#all beta -180 180
#all gamma -180 180
#all delta -180 180
#all epsilon -180 180
#all zeta -180 180
#all chi -180 180
#all p -180 180
#all vmax -180 180
#all v0 -180 180
#all v1 -180 180
#all v2 -180 180
#all v3 -180 180
#all v4 -180 180
#all eta -180 180
#all theta -180 180
|
To make limits for the required parameter(s) one should only remove "#" from the beginning of the
appropriate line and change the limits to the range of interest.
The following symbols / numbers are accepted when entered into the
limits entry box:
| type of contents | accepted symbols / numbers |
| residue number | 'all', the number of a single residue |
| parameter name | alpha, beta, gamma, delta, epsilon, zeta, chi, p, vmax, v0, v1, v2, v3, v4, eta, theta |
| min value | the number within the range <-180, 180> |
| max value | the number > min value and within the range <-180, 180> |
Search engine ignores:
- empty lines;
- comment lines, i.e. lines starting from "#",
if placed in the
limits entry box.
There is a possibility to load
basic query from user file into the
sequence / structure entry box. Such a file can be prepared by the user
in any text editor. It can be also created when one
saves the contents
of the
sequence / structure entry box.
The file should contain data in the
format accepted by RNA FRABASE.
Basic query entered into the
sequence / structure entry box can be saved to the text file
(click
Save button placed above the entry box). The default name for the file is
"basic_search.txt". Next, such a file can be used as an input, i.e. one can
upload the file
into the
sequence / structure entry box in any further session.
This menu allows very fast screening of basic structural elements, such as residues, base pairs, multiplets,
dinucleotide steps, stems, and single stranded regions of loops (apical, bulge, internal, and n-way junctions
(n ≤ 13)). In all cases, an instant visualisation of each fragment with JSmol is available.
Search of individual nucleotide residues can be based on the following filters:
- the residue; symbols encoding {any} residue or a ribonucleotide {A, C, G, U} or a modified
{a, c, g, u} residue can be selected
- the experimental method {any, X-Ray, NMR, Electron Microscopy, other}
- conformational parameters:
- sugar pucker pseudorotation phase angle (P),
- sugar pucker amplitude (Tm)
- glicosidic torsion angle (χ)
- exocyclic torsion angle around C4'-C5' (γ)
An option to include all models of the structure can be checked
Results are presented in the table; they can be saved by clicking the "Download results" button.
Moreover, information about coordinates in the PDB format, torsion angles and base pairs is obtained when
clicking respective buttons. One searching for a particular modified nucleotide residues is advised to use dedicated databases.
Search of canonical and non-canonical base pairs can be based on the following filters:
- residues; symbols encoding {any} residue, a ribonucleotide {A, C, G, U} or a modified
{a, c, g, u} residue can be selected
- the experimental method {any, X-Ray, NMR, Electron Microscopy, other}
- base-base parameters:
- shear (Sx),
- stretch (Sy),
- stagger (Sz),
- buckle (κ),
- propeller (ω),
- opening (δ)
An option to include all models of the structure can be checked.
Results are presented in the table; they can be saved by clicking the "Download results" button. Moreover,
information about coordinates in the PDB format and torsion angles is obtained when clicking respective buttons.
Multiplets were identified using RNAView software. They contain from 3 to 5 residues.
Search can be based on the following filters:
- residues; symbols encoding {any} residue, a ribonucleotide {A, C, G, U} or a modified
{a, c, g, u} residue can be selected
- the experimental method {any, X-Ray, NMR, Electron Microscopy, other}
An option to include all models of the structure can be checked.
Results are presented in the table; they can be saved by clicking the "Download results" button.
Moreover, information about coordinates in the PDB format, torsion angles and base pairs is obtained when
clicking respective buttons.
Search is applied to dinucleotide steps within two canonical base pairs.
Search can be based on the following filters:
- residues for the first and a second base pair; symbols encoding {any} residue, a ribonucleotide {A, C, G, U} or a modified {a, c, g, u} residue can be selected
- the experimental method {any, X-Ray, NMR, Electron Microscopy, other}
- Inter base pair parameters:
- shift (Dx)
- slide (Dy)
- rise (Dz)
- tilt (τ)
- roll (ρ)
- twist (Ω)
An option to include all models of the structure can be checked.
Results are presented in the table; they can be saved by clicking the "Download results" button. Moreover, information about coordinates in the PDB format, torsion angles and base pairs is obtained when clicking respective buttons.
Search is applied to duplex segments containing canonical base pairs.
Search can be based on the following filters:
- stem length; exact or interval length can be applied as an option;
- first strand sequence; a second strand is optional (all in the IUPAC notation);
- case sensitivity;
- the experimental method {any, X-Ray, NMR, Electron Microscopy, other}.
An option to include all models of the structure can be checked.
Results are presented in the table; they can be saved by clicking the "Download results" button.
Moreover, information about coordinates in the PDB format, torsion angles and base pairs is obtained when
clicking respective buttons.
The loop is defined as the construct composed of one strand (hairpin apical loops),
two strands (internal loops and bulges) and n-strands (n-way junctions) closed
by 1, 2 and n
canonical base pairs, respectively (see examples below).
Example 1
|
Example 2
|
Example 3
|
Example 4
|
The loop forming strand is defined by its length (i.e. by its length plus 2 residues). For example, a hairpin tetraloop should be defined
by the "length = 6". Its sequence is optional. Up to 13 strands can be defined.
Search can be based on the following filters:
An option to include all models of the structure can be checked.
Results are presented in the table; they can be saved by clicking the "Download results" button.
Moreover, information about coordinates in the PDB format, torsion angles and base pairs is obtained
when clicking respective buttons.
After clicking the
Search button a table with a list of all 3D RNA fragments which match the input data is presented.
The list contains the following information concerning each fragment:
- PDB Id of the whole structure (linked directly to the original PDB data),
- sequence,
- secondary structure in the dot-bracket notation,
- identifier of the chain, in which RNA fragment has been found,
- position of the fragment within the chain(s),
- a button to visualize a fragment of interest (JSmol)
- experimental method (as described in the appropriate PDB file),
- functional class (as described in the appropriate PDB file),
- date of structure deposition in PDB,
- structure resolution (as given in the appropriate PDB file).
- model of the structure
If the number of resulting fragments exceeds 50, they are displayed on several pages
(each contains the following 50 records).
The result table can be saved to the file (click
Save table) in CSV file format
(Comma Separated Values Standard File Format) which can be easily processed by different spreadsheet programs.
Default file name is "results_table.csv".
To see the conformational parameters one should select the fragment(s) of the interest (click the
appropriate check-box situated in the left-most column) and, next, click one the following buttons:
| Show coordinates |
|
click this button to see atom coordinates for selected fragment(s) |
|
| Show torsion angles |
|
click this button to see torsion angles, pseudorotation and sugar pucker amplitude for selected fragment(s) |
|
| Show base pairs |
|
click this button to see base pairs for selected fragment(s) |
Atom coordinates of RNA fragment(s) are displayed in the format used in PDB files. One can
save them directly to the file (in PDB format) by clicking
Save table. Default file name is "coordinates.pdb".
The atom coordinates data (file) are ready to be applied to the 3D RNA structure visualization programs. By clicking
Save table
a user may open the visualization program (if it is installed in a user's local diskspace) - atom coordinates are directly loaded into the program
and selected structure is displayed.
Displayed conformational parameters of the RNA fragment(s) can be saved to the file
(click
Save table) in CSV file format (Comma Separated Values Standard File Format)
compatible with spreadsheet programs. Default file name is "torsion_angles.csv".
Moreover, torsion angle statistics are now visualized after clicking
Show histogram button. Statistics for each parameter can be saved separately to the file (click
Save histogram) in CSV format.
Definition of torsion and pseudotorsion angles
| Angle | Atoms |
| alpha | O3'(n-1) - P - O5' - C5' |
| beta | P - O5' - C5' - C4' |
| gamma | O5' - C5' - C4' - C3' |
| delta | C5' - C4' - C3' - O3' |
| epsilon | C4' - C3' - O3' - P(n+1) |
| zeta | C3' - O3' - P(n+1) - O5'(n+1) |
chi (purines)
chi (pirymidines) | O4' - C1' - N9 - C4
O4' - C1' - N1 - C2 |
| v0 | C4' - O4' - C1' - C2' |
| v1 | O4' - C1' - C2' - C3' |
| v2 | C1' - C2' - C3' - C4' |
| v3 | C2' - C3' - C4' - O4' |
| v4 | C3' - C4' - O4' - C1' |
| eta | C4'(n-1) - P - C4' - P(n+1) |
| theta | P - C4' - P(n+1) - C4'(n+1) |
n - residue number
|
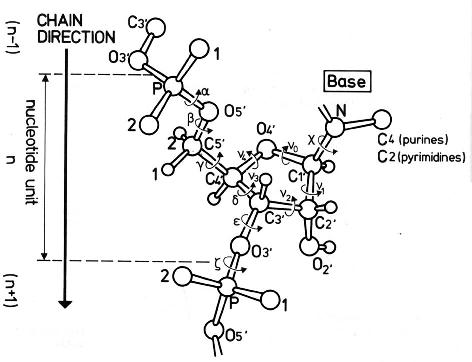
Saenger, W. (1984) Principles of Nucleic Acid Structure, Springer-Verlag.
|
Both canonical and non-canonical base pairs are classified according to the Westhof's and Saenger's
notation (
RNAView software) and their parameters, Shear (Sx), Stretch (Sy), Stagger (Sz), Buckle (κ),
Propeller (ω), Opening (δ) are calculated (
3DNA software).
Data can be saved to the file (click Save table) in CSV file format (Comma Separated Values Standard File Format)
compatible with spreadsheet programs. Default file name is "base_pairs.csv".
The term "intra base pair location" describes a situation where both residues are paired within the same fragment.
The term "inter base pair location" is for cases where only one residue is within the fragment of interest, the
second one is outside of this fragment. Both pieces of information are of help when searching for RNA fragments and motifs and
their interactions.
The detailed description of Westhof's and Saenger's classification is available here:
The table of a complete list of RNA secondary structures in the dot-bracket format is now available to the user.
Currently, the table contains entries derived from 2753 PDB files.
When pointing at the PDB id code of selected
RNA, an icon is popping up showing its 3D structure (based on PDB visualisation). An access to the PDB entry is
given.
Clicking the "show details" button gives the RNA sequence and its secondary structures in the dot-bracket
format. Each secondary structure can be now visualised in a graphic form.
The data can be saved as a text file using "save details" button.
Other table details reveal: number of
models, the functional class, experimental method used, its resolution, and the time of the RNA coordinates
deposition.
Popenda, M., Blazewicz, M., Szachniuk, M., Adamiak, R.W.
RNA FRABASE: an engine with database to search the three-dimensional fragments within 3D RNA structures,
Nucleic Acids Research 36, 2008, D386-D391
(published online on October 5, 2007, doi:
10.1093/nar/gkm786).
(please cite this paper if you use RNA FRABASE 1.0 in a published report)
Popenda, M., Szachniuk, M., Blazewicz, M., Wasik, S., Burke, E.K., Blazewicz, J., Adamiak, R.W.
RNA FRABASE 2.0: an advanced web-accessible database with the capacity to
search the three-dimensional fragments within RNA structures,
BMC Bioinformatics,
2010, 11:231
(published online on May 6, 2010, doi:
10.1186/1471-2105-11-231).
(please cite this paper if you use RNA FRABASE 2.0 in a published report)
